Sickle Cell Anaemia
Overview
Sickle cell anaemia is an autosomal recessive disorder
causing production of abnormal ß-globin chains. A single amino acid is
substituted in the ß-globin chain (Glu to Val at position 6). This results in
the production of HbS (haemoglobin Sickle) rather than HbA. The common variants
of sickle cell disease are:
- Sickle cell anaemia (SS disease) is the most common
- Sickle cell trait – causes no disability and protects from malaria except in hypoxia.
- Sickle ß Thalassemia (HbS/ßthal)
- Sickle haemoglobin C disease (HbSC)
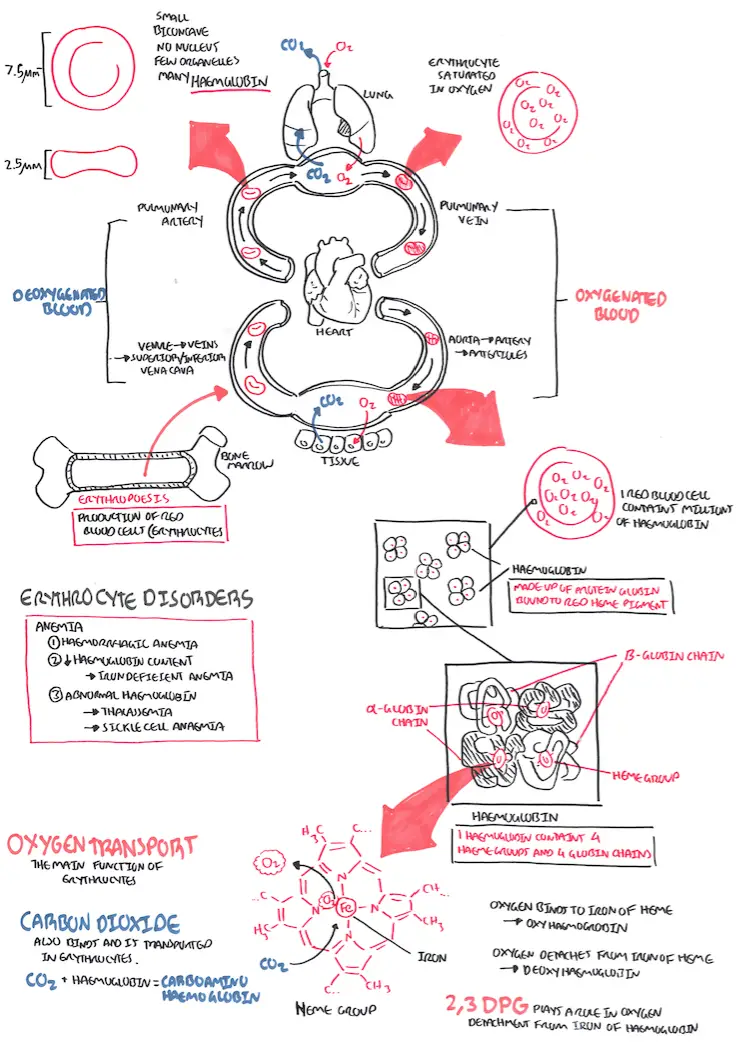
Red Blood Cells Physiology

Risk Factors
It is
hereditary! Genetic counselling and prenatal tests can help prevent.
Signs and Symptoms
Newborns are usually asymptomatic because babies still have
fetal haemoglobin
- Vaso-occlusive crisis
- Dactylitis (children)
- Mesenteric Ischaemia
- CNS infarction – seziures, stroke, cognitive defects
- Avascular necrosis (neck of femur)
- Leg ulcers
- Priapism
- Fever – infection
- Acute chest syndrome
- a new infiltrate on chest x-ray
- associated with one or more
NEW symptoms:
fever, cough, sputum production, dyspnea, or hypoxia. - Acute splenic sequestration
- Splenomegaly
- Hepatomegaly
- Aplastic crisis – due to parovirus infection, with a sudden reduction in bone marrow production
Remember children typically present with acute dactylitis. Males
can present with priapism
|
Differential Diagnosis
Differential
diagnosis of Haemolytic anaemia
- Autoimmune haemolysis
- Hereditary spherocytosis
- G6PD deficiency
Investigations
Sickle
cell disease can be diagnosed in newborns, as well as older persons, by
hemoglobin electrophoresis, isoelectric focusing, high-performance liquid
chromatography or DNA analysis
- FBC
- Blood smear – sickle cells and target cells
Remember Sickle cell trait have normal blood smear, sickle cell
anaemia does not!
|
Side note Target cells are found in Thalassaemia too.
|
- Sickle solubility test
- The parents of the affected child with sickle cell aneamia will show features of sickle cell trait.
Sickle
solubility test is where a mixture of Hb S in a
reducing solution such as sodium dithionite gives a turbid appearance
because of precipitation of Hb S, whereas normal Hb gives a clear solution.
|
Pathophysiology
The
substitution of one amino acid in the hemoglobin molecule results in sickle
hemoglobin. Amino acid changed from Glu to Val. As a result, RBCs sickle
in low oxygen states causing occlusion of blood vessels, increased viscosity,
and inflammation.
The
average life span of these sickle RBC are 20days (120days is normal)
Clinical
features
- Anaemia occurs due to splenic sequestration, bone marrow aplasia and further haemolysis.
- Vaso-occlusive crisis
- Dactylitis (children)
- Mesenteric Ischaemia
- CNS infarction – seziures, stroke, cognitive defects
- Avascular necrosis
- Leg ulcers
- Priapism
- Acute chest syndrome
- a new infiltrate on chest x-ray
- associated with one or more
NEW symptoms:
fever, cough, sputum production, dyspnea, or hypoxia. - Acute splenic sequestration
- A result of vaso-occulsion. The condition occurs in childhood before multiple infarctions have occurred. The latter eventually leads to a brotic non- functioning spleen. Splenomegaly and Hepatomegaly occur
- Aplastic crisis – This most commonly occurs following infection with parovirus B19, which invades proliferating erythroid progenitors. There is a rapid fall in haemoglobin with no reticulocytes in the peripheral blood, because of the failure of erythropoiesis in the marrow.
Management
Acute
crises may occur spontaneously, or may be
precipitated by:
- Infection
- Dehydration
- Hypoxia
- Sedatives, local anaesthetics and surgery
General
sickle crisis management
- FBE including reticulocyte count
- Blood group & cross match
- Blood and urine cultures if febrile
- IV Antibiotics
- EUC and LFT if jaundice or dehydrated
- Consider chest xray if febrile with respiratory symptoms
- Obtain CT without contrast to exclude bleed if concerns regarding stroke or MRI if available without delay
- Treatment
- Contact Haemtologist
- Analgesics (paracetamol or ibuprofen)
- Fluids(oral of IV)
- +/- blood transfusions (exchange transfusion)
Side note Hydroxyurea for prophylaxis of recurrent sickle cell
crisis. Eventually person will need regular vaccination because of spleen
problems
|
Acute
Cell Crisis can present with the following
- Vaso-occlusive crisis
- Stroke
- Priapism
- Fever
- Acute chest syndrome
- Acute splenic sequestration
- Aplastic crisis
Complications and Prognosis
Complication
- Splenic infarction (<2yo)
- Increase risk of infection
- Failure to thrive
- Chronic renal failure
- Gallstone
- Iron overload
- Lung damage – Hypoxia → fibrosis → pulmonary hypertension
- Aplastic crisis – Paravirus B19 infection causing drop haemotocrit
Remember Paravirus B19 infection causing drop haemotocrit in
sickle cell and thalassaemia. Treatment is immunoglobulins
|
Lê Văn Công
Reference: Armando Hasudungan

